

DeepMind AlphaFold for antibody discovery: What's the status?
 10 min read
10 min read- Quick recap: What is AlphaFold and how does it work?
- Can researchers use AlphaFold for antibody discovery?
- Benchmarking AF2-multimer to distinguish antibody/nanobody binders and non-binders against diverse antigens
- Benchmarking AF2-multimer to distinguish binders from non-binders against a single antigen
- Experimental conclusion
- The future of AlphaFold
- NaturalAntibody develops bioinformatics solutions that facilitate the process of discovering antibody-based therapeutics. We use AI-based computational methods to support researchers in making the process of designing therapeutic antibodies faster and safer.
- To learn more,
There are over 100 million known proteins, and many more are discovered every year. Each protein has a unique 3D shape that determines how it works and what it does. But discovering the structure of these proteins remains an expensive and often time-consuming process, so we only know the structure of a tiny fraction of the proteins known to science.
Developing new methods to quickly predict these structures of millions of unknown proteins could help us tackle diseases and find new drugs faster. This is the problem addressed by AlphaFold2 (AF2), a solution developed by DeepMind, a company belonging to Alphabet/Google, revealed in a seminal Nature paper.
AF2 is based on a machine learning approach that incorporates both physical and biological knowledge into the design of its deep learning algorithms.
But can this piece of cutting-edge software help in the antibody discovery process? Our team at NaturalAntibody carried out some tests to check its potential. Read on to find out what we discovered.
Quick recap: What is AlphaFold and how does it work?
The basic building blocks of proteins are amino acids. The attraction and repulsion between the amino acids cause the chain to fold into a 3D structure, which determines the function of a protein.
The protein folding problem scientists have been trying to solve for many years focuses on discovering the link between the string of amino acids and the protein conformation. Experimental techniques are laborious and time-consuming, sometimes taking years and millions of dollars. For instance, it was estimated that reproducing the contents of the Protein Data Bank, which curates known 3D structures of macromolecules, would cost some $12 billion.
AlphaFold2 can predict the shape of a protein at scale in just a few minutes, showing the incredible potential of AI approaches to science. The solution was trained on sequences and structures of around 100,000 known proteins.
AlphaFold Protein Structure Database, created in partnership with Europe's flagship laboratory for life sciences (EMBL's European Bioinformatics Institute), is a comprehensive reference database representing 350,000 structures, including the human proteome (all of the ~20,000 known proteins expressed in the human body) along with the proteomes of 20 additional organisms important for biological research.
The human proteome is among the most important biological discoveries of recent decades. Its release has significantly expanded our knowledge of protein structures, more than doubling the number of high-accuracy protein structures available to scientists around the world.
Can researchers use AlphaFold for antibody discovery?
There's no denying that AlphaFold is an excellent tool for solving protein structures. The other “unsolved” problem in bioinformatics is protein-protein binding. Antibody-antigen recognition is a sub-problem of that.
Previous studies showed that AlphaFold2 confidence scores (pIDDT) are not a bad proxy for calling protein-protein interactions (Bryant, Pozzati, and Elofsson 2022). This raises hope for employing AF2 in antibody discovery - distinguishing across multiple (potentially millions) antibodies to identify those that could bind a target in question.
To address this issue, we tested the ability of AF2-multimer (the next version of AF2 to tackle ‘multimers’) (Evans et al. 2022) to distinguish correct and incorrect antibody-antigen pairs.
We did so by setting AF2 against the challenging task of giving it the sequence of both the antibody and antigen (correct and incorrect pairs) and checking if the confidence scores (pIDDT) were better in the correct configuration (Figure 1).
To test AlphaFold, we ran two experiments. The first one checked its ability to distinguish interacting antibody/nanobody protein pairs across a wide range of antigens. The second experiment tested AlphaFold’s ability to distinguish correct binding pairs from incorrect ones from a set of multiple binders and non-binders against a single antigen.
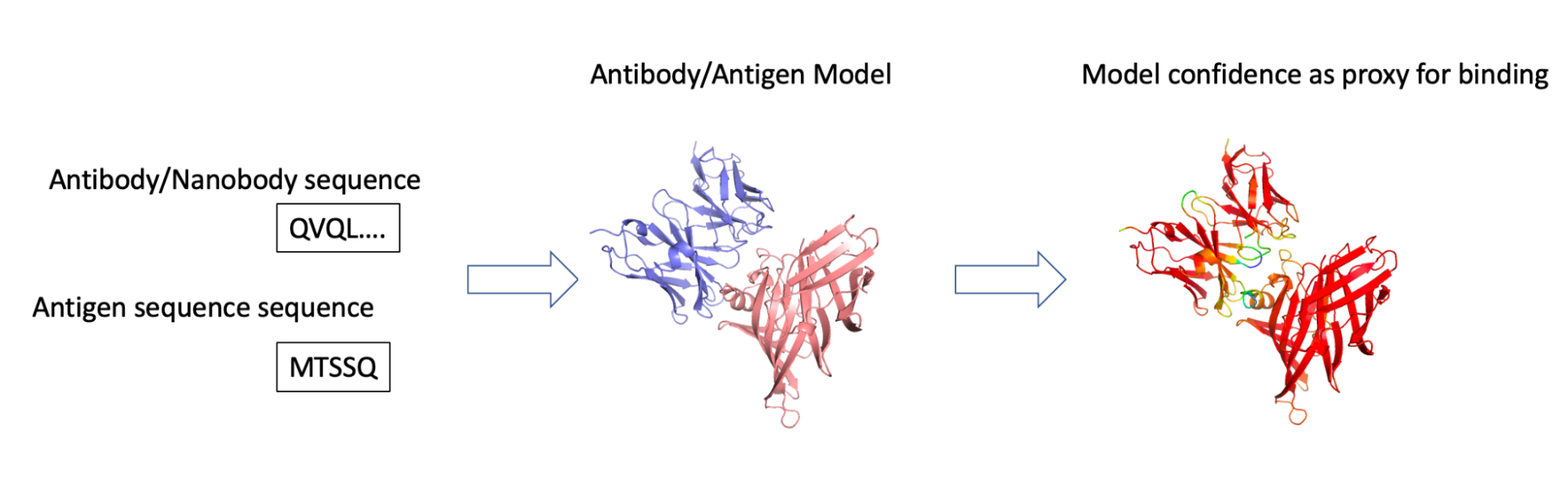
Figure 1. We employed our federated antibody database to identify pairs of sequences of interacting antibodies/antigens. These were fed into AF2-multimer, and the pIDDT model confidence score was used as an indicator of whether the two could indeed interact.
Benchmarking AF2-multimer to distinguish antibody/nanobody binders and non-binders against diverse antigens
We employed our database of curated antibody/nanobody-target sequences to devise a dataset of binders (positive set) and non-binders (negative set).
For binders, we employed a set of antibodies against targets smaller than 200-amino-acids-long, just to go easier on AF2 having to model larger proteins. As the negative set, we randomly assigned the antibodies/nanobodies from the positive set to incorrect antigens.
Though a certain degree of polyspecificity is to be expected, it’s quite unlikely that a large portion of shuffled molecules would bind the “false” target. For the antibody dataset, such a procedure resulted in 45 antibody-antigen pairs and 28 for nanobodies.
As the metric of confidence of calling a binder, we employed the pIDDT that gives the confidence of coordinates. Such a score was previously reported as a proxy to make protein interaction calls (Johansson-Åkhe and Wallner 2021). We made three versions of the score:
- antibody_score - the average pIDDT for the antibody portion (paratope, CDRs).
- antigen_score - the average pIDDT for the antigen portion (epitope).
- combined_score - a combination of both.
We broke the scores up since it wasn’t clear a priori what the best way to compare the scores of diverse antibodies against a single antigen is. The results of running the three scores on the antibody and nanobody datasets are shown in Figure 2.
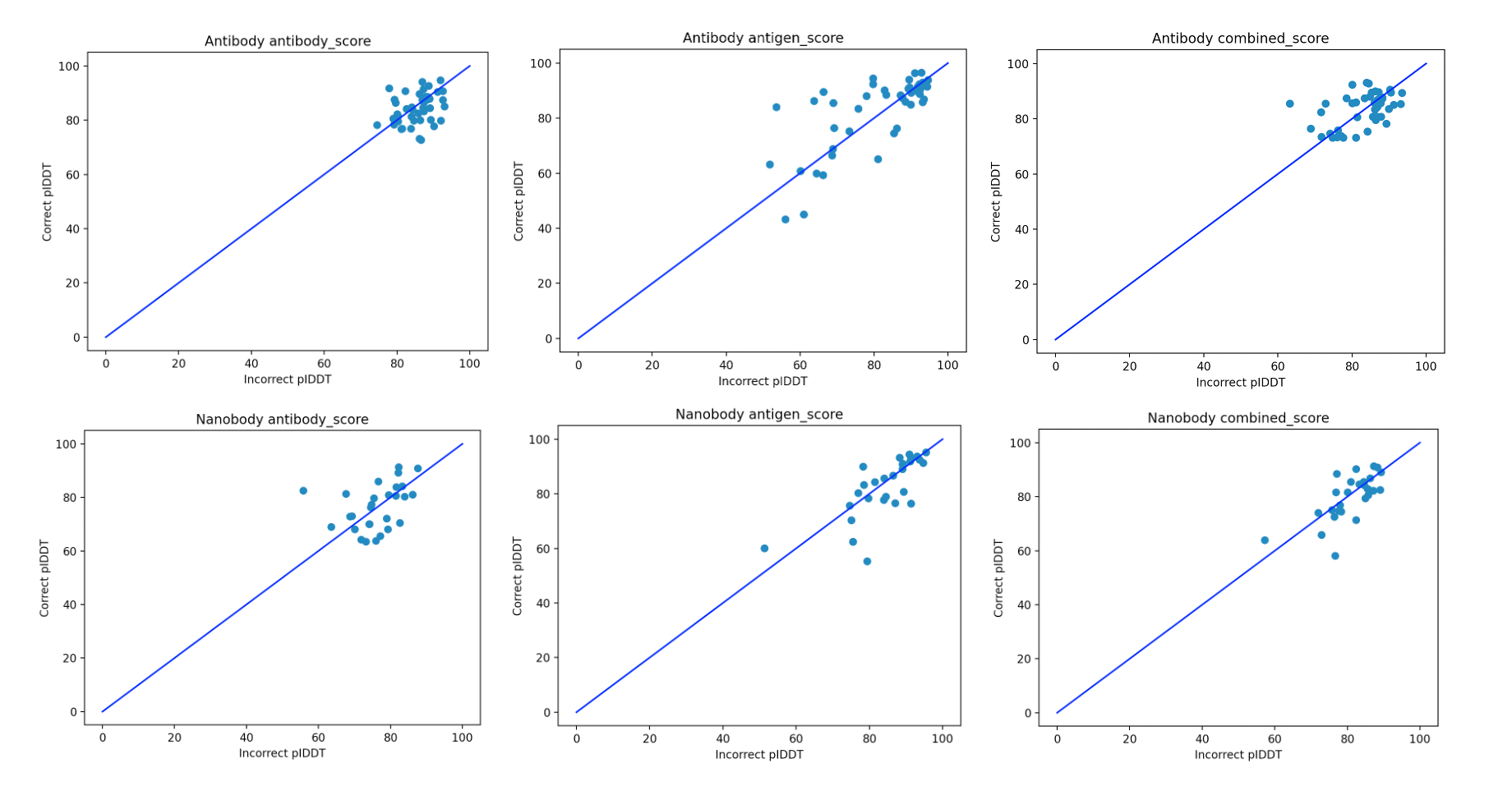
Figure 2. Contrasting the pIDDT scores for the correct/incorrect pairs of antibody/nanobody antigens for the three scores for which we checked the predictive power.
The results in Figure 2 indicate that there doesn’t appear to be a clear separation of the positive and negative antibodies against diverse targets, which is further qualified by Table 1. As the problem is of the binary distinction between two potential binders against a single antigen, a random/non-informative model is expected to produce accuracy close to 50%. In all of the cases, all the scores are in this range. This suggests that such basic use of AF2-multimer doesn’t provide a solution to distinguishing binders from non-binders.
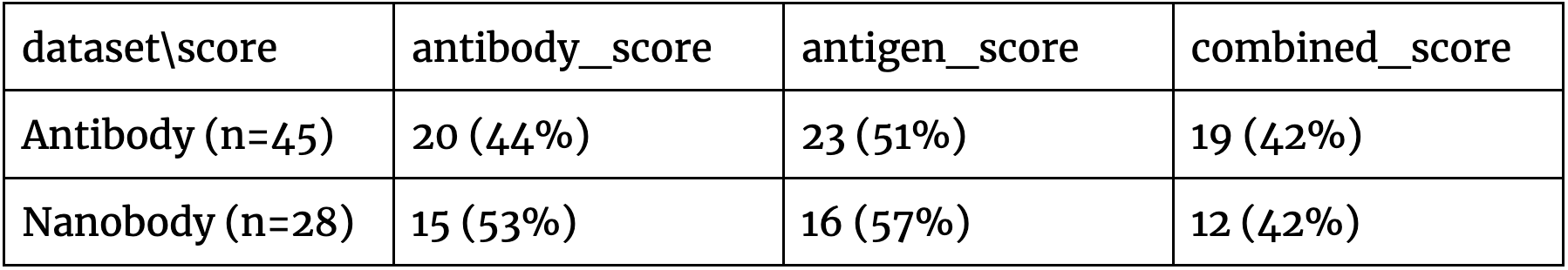
Table 1. The proportion of the pairs where the correct antibody/nanobody-antigen assignment had a higher pIDDT score than the incorrect one. In all cases, the predictions don’t appear strong as the baseline for this predictive task is 50% accuracy.
Benchmarking AF2-multimer to distinguish binders from non-binders against a single antigen
A typical antibody discovery task is to filter out a set of antigen-specific antibodies from a set containing (hopefully) binders and (likely) non-binders (Trück et al. 2015). To check AF2-multimer’s ability to tackle this task, we employed a previous set of binders and non-binders against OVA from a previous study (Goldstein et al. 2020).
Corrects: Only the proteins that were reported as binding were used for testing as the positive set, removing one reported as non-expressing and with no detected binding. The four non-binding molecules were tested and labeled that way. However, one of them failed to produce AF2 output (rIGHV5-34_rIGKV12S30).
Incorrects: As the negative set, we performed a similar experiment to the one carried out by the authors in the publication. They shuffled the heavy and light chains. Most of the chains expressed but didn’t have OVA binding anymore (86 expressed and only five showed OVA reactivity). So, we randomly shuffled chains of the positive set and labeled such antibodies as the negative set.
Altogether, we were able to produce AF2-multimer scores for 74 positive molecules and 90 negative molecules. We randomly downsampled the negative set to 74 molecules to balance the test set.
We employed an OVA sequence from Uniprot P01012 as the target antigen.
Unlike in the previous section, where we faced a binary binder/non-binder problem, here we wished to filter out binders from non-binders from a larger set. For a given score (e.g., average antibody_score > 90), we count the number of correct and incorrect binders above it. This aims to simulate a situation of us actually picking the binders for high pIDDT scores. If the method works, one would expect to have a higher enrichment in actual binders. As a control, we also shuffled the correct/incorrect scores to get a control baseline.
The results of this scheme for our three pIDDT variations are given in Figure 3. Though the enrichment of binders always appears higher than non-binders, it doesn’t go beyond the standard deviation for the baseline randomly shuffled control. Unfortunately, according to this experiment, the basic pIDDT scores weren’t predictive of the antibody-antigen interactions.
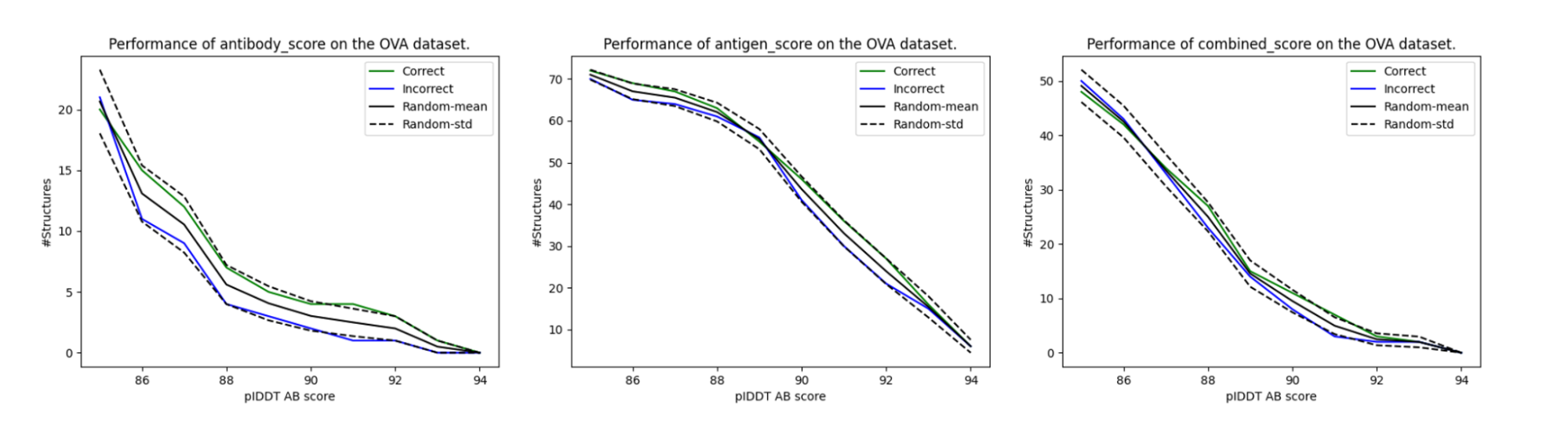
Figure 3. Enrichment of the correct and incorrect assignments by highest pIDDT scores. Though the correct assignments appear to have higher enrichment in the graph than the incorrect assignments, they are within the standard error with respect to the random assignments of the scores.
Experimental conclusion
We performed a very easy experiment to check if there’s “a free lunch” for antibody discovery employing AlphaFold2. Unfortunately, according to our results, this is not the case. That’s not entirely surprising and confirms earlier results (Yin et al. 2021).
AF2 leverages evolutionary information, which is a distinguishing feature of protein structure as well as protein-protein interactions. But antibodies follow different binding preferences than normal protein-protein interactions (Krawczyk et al. 2013). Since methods such as AF2 don’t appear to learn the underlying physics (Outeiral, Nissley, and Deane 2022), but rather the probability distribution/statistics, if antibodies follow a different statistic (as chemistry is the same!), it’s not entirely surprising that this approach doesn’t work.
Still, this doesn’t mean that a much more nuanced approach wouldn’t be able to leverage AF2 in some way to tackle the problem of antibody-antigen binding.
The future of AlphaFold
Researchers around the world are using AF2 to make strides in areas that are important to society. For instance, the Centre for Enzyme Innovation at the University of Portsmouth (CEI) is using AlphaFold's predictions to help engineer faster enzymes for recycling some of our most polluting single-use plastics.
A team at the University of Colorado Boulder is finding promise in using AlphaFold's predictions to study antibiotic resistance, while a group at the University of California San Francisco has used them to increase their understanding of SARS-CoV-2 biology.
Specifically in the antibody world, the large amount of information made available by modeling known proteins in the AlphaFold Database (Varadi et al. 2022), made available a whole set of potential targets that antibodies could tackle.
If protein structure prediction is possible to solve by clever usage of data that was out there all along, it’s plausible to think that the cognate binding problem could be solved as well.
Such applications into the antibody world would open a whole new chapter in the way we discover novel therapeutics by speeding up the process by orders of magnitude.
___
NaturalAntibody develops bioinformatics solutions that facilitate the process of discovering antibody-based therapeutics. We use AI-based computational methods to support researchers in making the process of designing therapeutic antibodies faster and safer.
To learn more, book a 30-minute demo with Dr. Konrad Krawczyk.
___
References:
Bryant, Patrick, Gabriele Pozzati, and Arne Elofsson. 2022. “Improved Prediction of Protein-Protein Interactions Using AlphaFold2.” Nature Communications 13 (1): 1265.
Evans, Richard, Michael O’Neill, Alexander Pritzel, Natasha Antropova, Andrew Senior, Tim Green, Augustin Žídek, et al. 2022. “Protein Complex Prediction with AlphaFold-Multimer.” bioRxiv. https://doi.org/10.1101/2021.10.04.463034.
Johansson-Åkhe, Isak, and Björn Wallner. 2021. “Benchmarking Peptide-Protein Docking and Interaction Prediction with AlphaFold-Multimer.” bioRxiv. https://doi.org/10.1101/2021.11.16.468810.
Krawczyk, Konrad, Terry Baker, Jiye Shi, and Charlotte M. Deane. 2013. “Antibody I-Patch Prediction of the Antibody Binding Site Improves Rigid Local Antibody–antigen Docking.” Protein Engineering, Design & Selection: PEDS 26 (10): 621–29.
Outeiral, Carlos, Daniel A. Nissley, and Charlotte M. Deane. 2022. “Current Structure Predictors Are Not Learning the Physics of Protein Folding.” Bioinformatics, January. https://doi.org/10.1093/bioinformatics/btab881.
Goldstein, L.D., Chen, YJ.J., Wu, J. et al. Massively parallel single-cell B-cell receptor sequencing enables rapid discovery of diverse antigen-reactive antibodies. Commun Biol 2, 304 (2019). https://doi.org/10.1038/s42003-019-0551-y
Trück, Johannes, Maheshi N. Ramasamy, Jacob D. Galson, Richard Rance, Julian Parkhill, Gerton Lunter, Andrew J. Pollard, and Dominic F. Kelly. 2015. “Identification of Antigen-Specific B Cell Receptor Sequences Using Public Repertoire Analysis.” Journal of Immunology 194 (1): 252–61.
Varadi, Mihaly, Stephen Anyango, Mandar Deshpande, Sreenath Nair, Cindy Natassia, Galabina Yordanova, David Yuan, et al. 2022. “AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models.” Nucleic Acids Research 50 (D1): D439–44.
Yin, Rui, Brandon Y. Feng, Amitabh Varshney, and Brian G. Pierce. 2021. “Benchmarking AlphaFold for Protein Complex Modeling Reveals Accuracy Determinants.” bioRxiv. https://doi.org/10.1101/2021.10.23.465575.