


Machine learning filtering of antibody NGS: NaturalAntibody Batch Sequence Annotation module
 10 min read
10 min readThe development of successful antibody-based therapeutics relies on meeting a range of biophysical properties, such as solubility, aggregation, expression, etc. - collectively known as ‘developability’ (Jain et al. 2017). Performing an array of experimental assays is typically constrained to a more select set of candidates at later stages, as analyzing thousands or even millions of molecules from a phage display experiment or animal immunization is still prohibitive cost- and time-wise.
To boost the chance of molecules with the best potential developability properties making it to the clinic, teams use computational methods that filter out the most promising molecules at the earliest possible stage (Khetan et al. 2022). Simple selection methods, such as clustering or basic post-translational liability detection, cede way to more advanced, statistical, and structure-driven methods (Raybould et al. 2019).
To meet the criteria of performing NGS filtering in the discovery process, the computational method needs to be:
- Accurate in offering developability predictions concerning experimental methods.
- Fast to be able to process millions of NGS reads in a reasonable time, to offer complementarity to the low-throughput experimental methods.
At NaturalAntibody, we developed a module called Batch Sequence Annotation. Here, users can upload a set (batch) of sequences and filter them by machine learning attributes to rapidly weed out molecules that might be a liability risk. The models are developed employing machine learning methods using copious antibody-specific information found in NaturalAntibody databases. The predictions are state-of-the-art accurate and engineered to produce results fast, making NGS-based filtering on a million-sequence scale or more possible.
The engine behind Batch Sequence Annotation
Our Batch Sequence Annotation is an interface for several machine learning annotations, such as immunogenicity and developability scores. The scores are the following, and they indicate:
- Immunogenicity. Our immunogenicity predictor correlated with the Anti-Drug Antibody (ADA) scores (Vaisman-Mentesh et al. 2019).
- Two Structural Developability Scores. Computational/structural developability guidelines based on a set of clinical stage antibodies first introduced in Raybould et al. 2019.
The first score is neural networks trained on large-scale NaturalAntibody data that we have trained based on the literature best practices. Our immunogenicity prediction achieves r2 in the range of 0.2 – 0.4 correlates with ADA scores that fall within state-of-the-art (Marks et al. 2021; Prihoda et al. 2022; Leem et al. 2022).
For our developability scores, we build on the best practices introduced by the Therapeutic Antibody Profiler (TAP) (Raybould et al. 2019). TAP is now widely used to offer indications of molecules with favorable developability properties (Mason et al. 2021; Bachas et al. 2022). Rather than focusing on predicting separate scores for developability assays (e.g., aggregation (Jain, Boland, et al. 2017), solubility (Sormanni, Aprile, and Vendruscolo 2015)), it derives a range of sequence/structural scores from therapeutic antibodies that passed the manufacturing hoops and uses these as reference for your query sequence. The main issue with this method was its speed, as it required an antibody structure prediction for some of its scores.
Benchmarking our approach
We have benchmarked the speed of the original method based on the standard homology procedure of finding the template from structures in the PDB (Krawczyk et al. 2018), modeling loops with FREAD (Choi and Deane 2010), packing heavy/light using ABAngle (Dunbar et al. 2013) and predicting side chains using PEARS (Leem et al. 2018). These methods are excellent (shameless founder bias) and freely available to the scientific community, and a protocol outlining collating them together is described in ABodyBuilder (Leem et al. 2016), which can achieve structure prediction in the order of one minute.
One can derive the calculations directly from sequence and structure by employing such a basic structure prediction tool. Benchmarking the speed of the tool on 100,000 paired sequences from the Observed Antibody Space database, we achieved a mean of 55.6 seconds per molecule. Back-of-the-envelope calculation puts the running time on 100,000 sequences on one core at roughly 1,544 hours or 64 days. Putting moar (sic) CPUs to the task can surely reduce the runtime, but it is still a bit long, so the method in the basic form requires some computational tuning to be fast enough for reliable NGS analyses.
Much of the lag in the method is taken by structural modeling. Advances in protein structure prediction by AlphaFold inspire deep learning antibody structure predictions (Ruffolo et al. 2022; Abanades et al. 2022). These produce the predictions' order of magnitude faster, within seconds. One aside, the neural networks can produce ‘unrefined’ structures with unrealistic bond geometries fast, which then require energetic minimization to produce a pristine structure. For applications such as TAP, one needs good-quality structures.
For simplicity, let’s assume that one replaced the homology structural modeling in TAP with deep learning and further assume that the entire calculation now takes around one second with GPU support. In such an optimistic scenario, the calculation would take roughly 27.7 hours for around 100,000 sequences. That is already better; however, with the amount of sequencing data from NGS in order of magnitude millions, a similar annotation of 1,000,000 sequences would take roughly 277 hours, meaning 11 days, which is quite a lot of GPU hours that also translate to costs.
At NaturalAntibody, we also have a deep learning method to predict the structure of antibodies (who doesn’t these days?). However, we wanted to engineer the entire pipeline of structural prediction -> developability scores to further bring down the running time. For this reason, we have engineered a unified structure prediction and developability score calculation framework. If deployed on a GPU instance with a bit of NaturalAntibody tinkering, it can achieve speeds of annotation of up to 200 molecules per second. This translates to roughly 720,000 antibody predictions per hour.
This is why our solution achieves state-of-the-art accuracy for a protocol, but with a bit of NaturalAntibody engineering, it can be deployed at scale, required by current NGS outputs for ML-augmented discovery campaigns. The method can be deployed in-house per discussion with NaturalAntibody or can be accessed via the NaturalAntobody platform as shown below.
Batch Sequence Annotation GUI
Batch Sequence Annotation is a module available in the NaturalAnitbody platform that lets users upload a set (batch) of antibody sequences in a well-adopted format (e.g., .fastq or .fasta) and filter it by computational/machine learning attributes such as immunogenicity or hydrophobicity in CDR regions or in the structural Fv charge symmetry parameters.
With Batch Sequence Annotation you can:
- Annotate multiple sequences with statistical/machine learning scores to get a data-driven bird's eye view of machine learning scores in your dataset.
- Study “Sequence Details” to analyze your chosen sequence in the Sequence Engineering module in more detail.
- Filter analysis results by sequence ID or statistical/machine learning scores to remove molecules predicted to have developability risks.
How does Batch Sequence Annotation work?
The first step is uploading a batch of antibody sequences. The platform supports .fastq and .fasta formats, with customization possible.
After uploading your sequences, you can choose which machine learning score to include in the analysis.
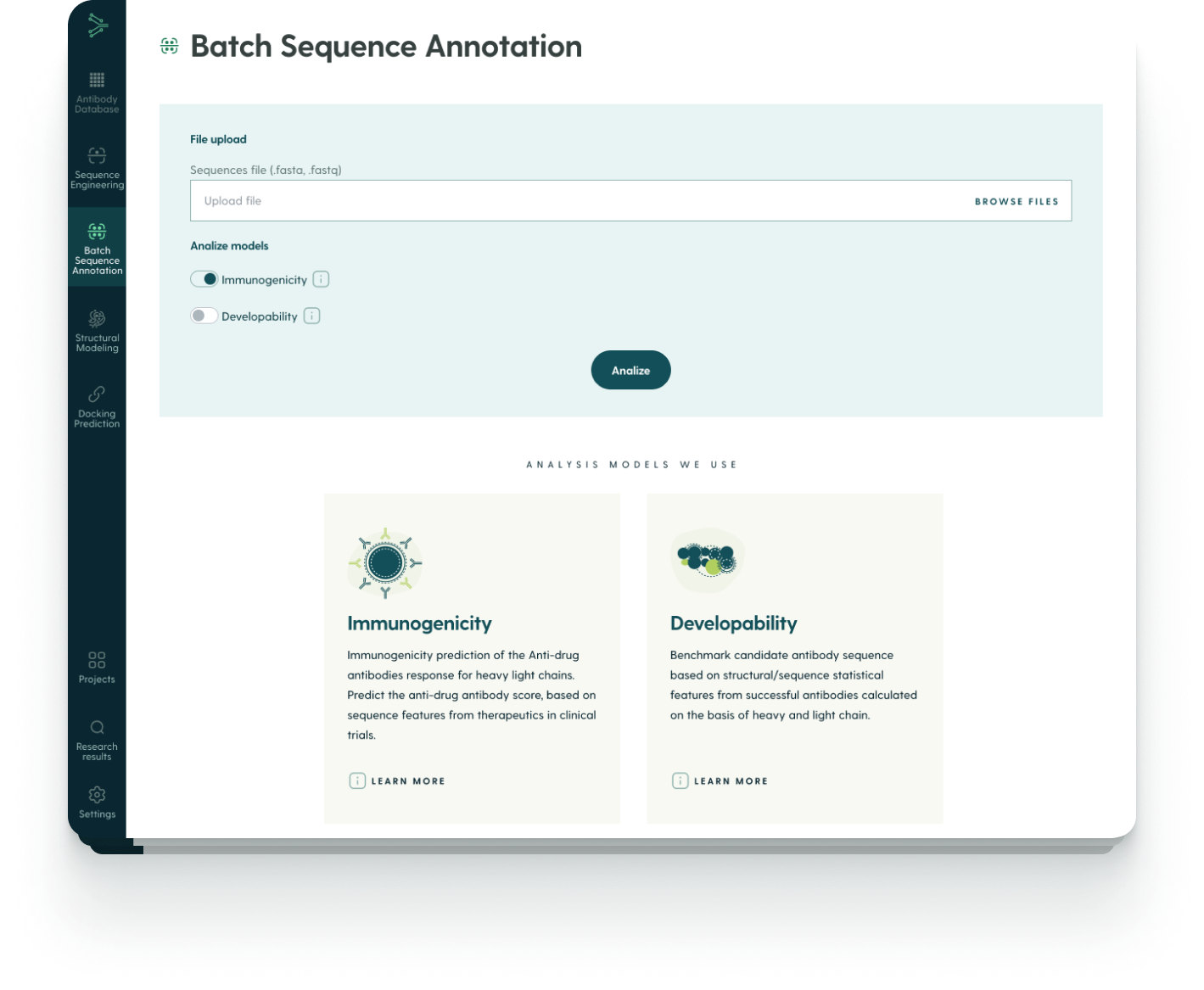
After you click on Browse Files, the platform analyzes your file in the context of the prediction models you picked.
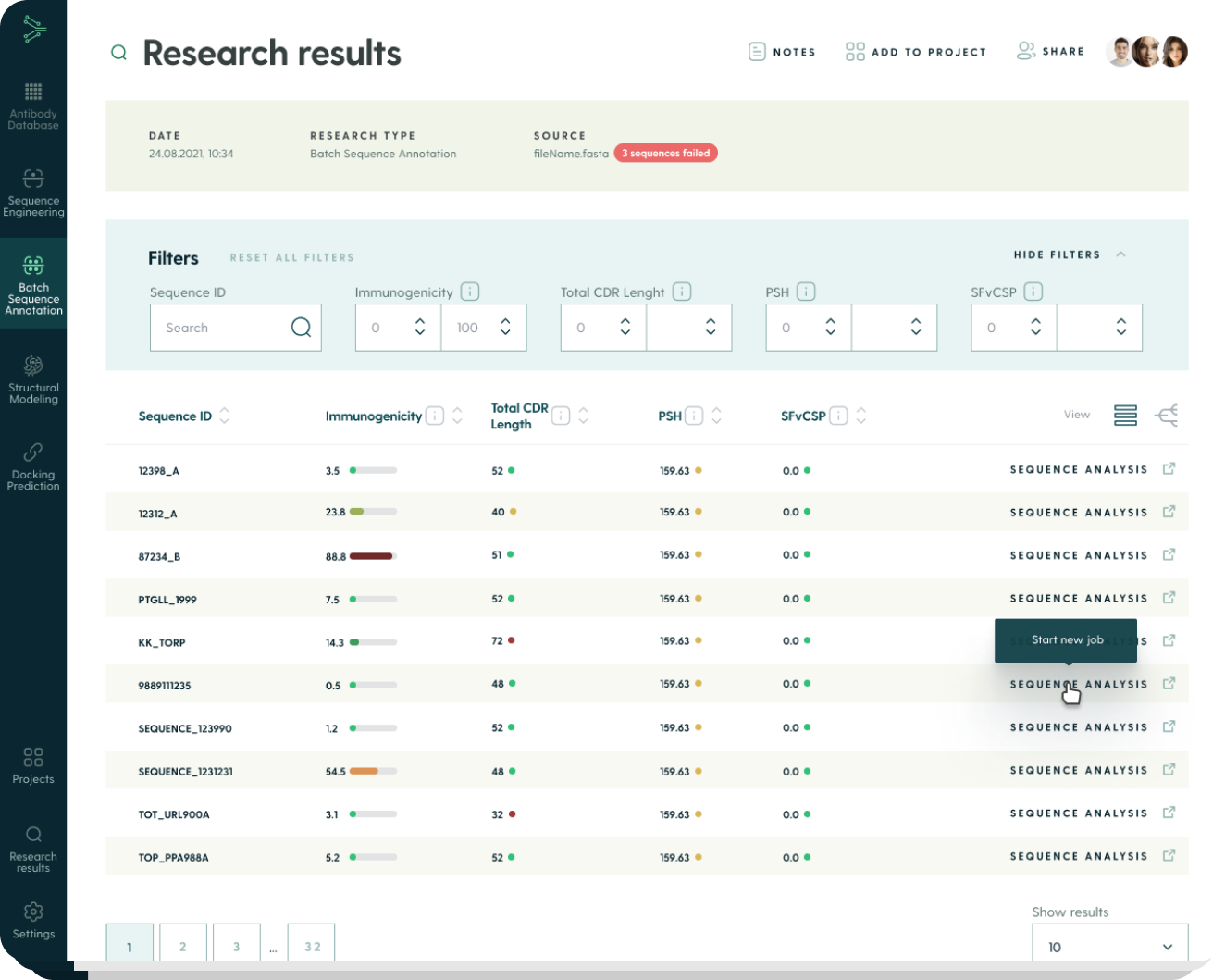
You receive research results with annotated sequences. You can filter and sort them using statistical/machine learning scores to find those predicted to be the most promising for your research project.
The platform allows you to narrow down a very long list of sequences into a few items for further research.
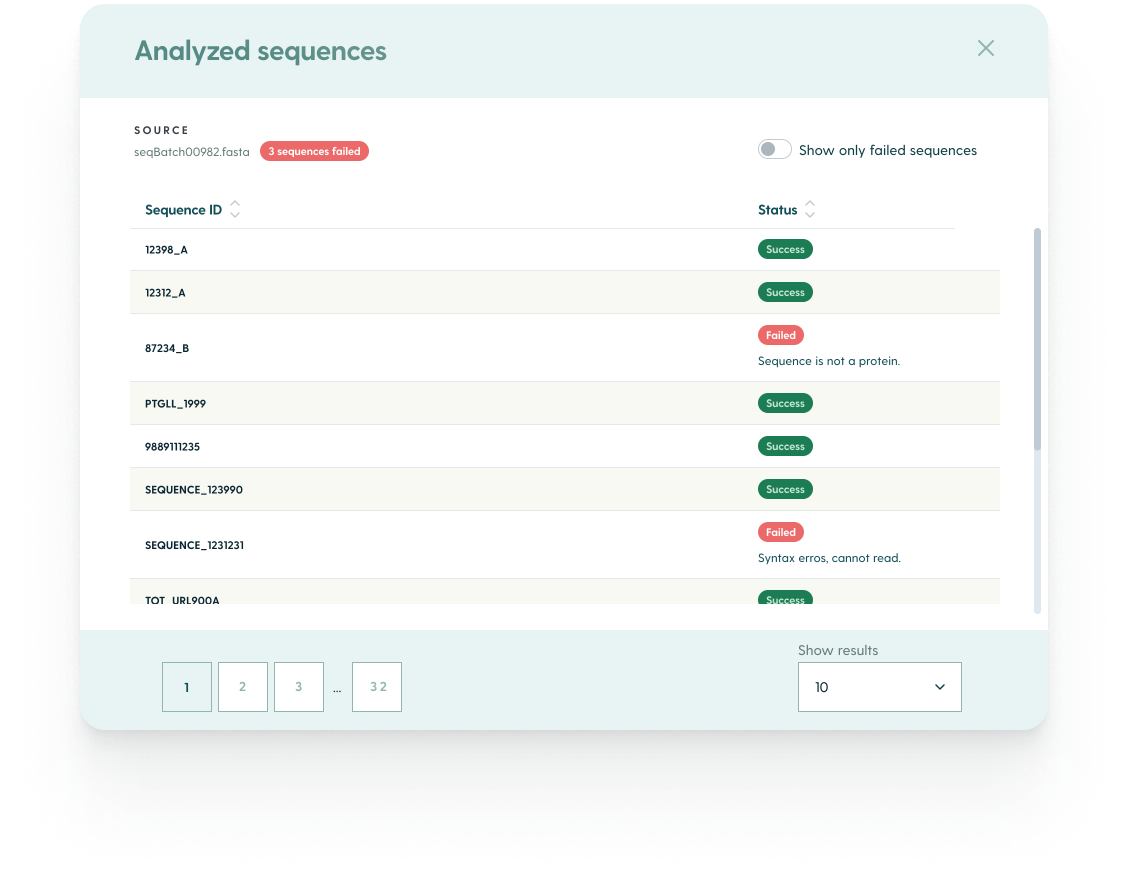
The module includes a detailed report about the analyzed sequences. You can check it by clicking on Source at the top of your research results list. You will see which sequences weren’t analyzed for specific predictions - and why (e.g., trivial sequence issues making them unable to be recognized as a valid antibody sequence).
Schedule a demo to see how the Batch Sequence Annotation module works in a real-world example.
________
References:
Abanades, Brennan, Guy Georges, Alexander Bujotzek, and Charlotte M. Deane. 2022. “ABlooper: Fast Accurate Antibody CDR Loop Structure Prediction with Accuracy Estimation.” Bioinformatics, January. https://doi.org/10.1093/bioinformatics/btac016.
Bachas, Sharrol, Goran Rakocevic, David Spencer, Anand V. Sastry, Robel Haile, John M. Sutton, George Kasun, et al. 2022. “Antibody Optimization Enabled by Artificial Intelligence Predictions of Binding Affinity and Naturalness.” bioRxiv. https://doi.org/10.1101/2022.08.16.504181.
Choi, Yoonjoo, and Charlotte M. Deane. 2010. “FREAD Revisited: Accurate Loop Structure Prediction Using a Database Search Algorithm.” Proteins 78 (6): 1431–40.
Dunbar, James, Angelika Fuchs, Jiye Shi, and Charlotte M. Deane. 2013. “ABangle: Characterising the VH--VL Orientation in Antibodies.” Protein Engineering, Design & Selection: PEDS 26 (10): 611–20.
Jain, Tushar, Todd Boland, Asparouh Lilov, Irina Burnina, Michael Brown, Yingda Xu, and Maximiliano Vásquez. 2017. “Prediction of Delayed Retention of Antibodies in Hydrophobic Interaction Chromatography from Sequence Using Machine Learning.” Bioinformatics 33 (23): 3758–66.
Jain, Tushar, Tingwan Sun, Stéphanie Durand, Amy Hall, Nga Rewa Houston, Juergen H. Nett, Beth Sharkey, et al. 2017. “Biophysical Properties of the Clinical-Stage Antibody Landscape.” Proceedings of the National Academy of Sciences of the United States of America 114 (5): 944–49.
Khetan, Rahul, Robin Curtis, Charlotte M. Deane, Johannes Thorling Hadsund, Uddipan Kar, Konrad Krawczyk, Daisuke Kuroda, et al. 2022. “Current Advances in Biopharmaceutical Informatics: Guidelines, Impact and Challenges in the Computational Developability Assessment of Antibody Therapeutics.” mAbs 14 (1): 2020082.
Krawczyk, Konrad, Sebastian Kelm, Aleksandr Kovaltsuk, Jacob D. Galson, Dominic Kelly, Johannes Trück, Cristian Regep, et al. 2018. “Structurally Mapping Antibody Repertoires.” Frontiers in Immunology 9 (July): 1698.
Leem, Jinwoo, James Dunbar, Guy Georges, Jiye Shi, and Charlotte M. Deane. 2016. “ABodyBuilder: Automated Antibody Structure Prediction with Data–driven Accuracy Estimation.” mAbs 8 (7): 1259–68.
Leem, Jinwoo, Guy Georges, Jiye Shi, and Charlotte M. Deane. 2018. “Antibody Side Chain Conformations Are Position-Dependent.” Proteins 86 (4): 383–92.
Leem, Jinwoo, Laura S. Mitchell, James H. R. Farmery, Justin Barton, and Jacob D. Galson. 2022. “Deciphering the Language of Antibodies Using Self-Supervised Learning.” Patterns of Prejudice, May, 100513.
Marks, Claire, Alissa M. Hummer, Mark Chin, and Charlotte M. Deane. 2021. “Humanization of Antibodies Using a Machine Learning Approach on Large-Scale Repertoire Data.” Bioinformatics, June. https://doi.org/10.1093/bioinformatics/btab434.
Mason, Derek M., Simon Friedensohn, Cédric R. Weber, Christian Jordi, Bastian Wagner, Simon M. Meng, Roy A. Ehling, et al. 2021. “Optimization of Therapeutic Antibodies by Predicting Antigen Specificity from Antibody Sequence via Deep Learning.” Nature Biomedical Engineering 5 (6): 600–612.
Młokosiewicz, Jakub, Piotr Deszyński, Wiktoria Wilman, Igor Jaszczyszyn, Rajkumar Ganesan, Aleksandr Kovaltsuk, Jinwoo Leem, Jacob Galson, and Konrad Krawczyk. 2021. “AbDiver – A Tool to Explore the Natural Antibody Landscape to Aid Therapeutic Design.” bioRxiv. https://doi.org/10.1101/2021.11.03.467080.
Prihoda, David, Jad Maamary, Andrew Waight, Veronica Juan, Laurence Fayadat-Dilman, Daniel Svozil, and Danny A. Bitton. 2022. “BioPhi: A Platform for Antibody Design, Humanization, and Humanness Evaluation Based on Natural Antibody Repertoires and Deep Learning.” mAbs 14 (1): 2020203.
Raybould, Matthew I. J., Claire Marks, Konrad Krawczyk, Bruck Taddese, Jaroslaw Nowak, Alan P. Lewis, Alexander Bujotzek, Jiye Shi, and Charlotte M. Deane. 2019. “Five Computational Developability Guidelines for Therapeutic Antibody Profiling.” Proceedings of the National Academy of Sciences of the United States of America 116 (10): 4025–30.
Ruffolo, Jeffrey A., Lee-Shin Chu, Sai Pooja Mahajan, and Jeffrey J. Gray. 2022. “Fast, Accurate Antibody Structure Prediction from Deep Learning on Massive Set of Natural Antibodies.” bioRxiv. https://doi.org/10.1101/2022.04.20.488972.
Sormanni, Pietro, Francesco A. Aprile, and Michele Vendruscolo. 2015. “The CamSol Method of Rational Design of Protein Mutants with Enhanced Solubility.” Journal of Molecular Biology 427 (2): 478–90.
Vaisman-Mentesh, Anna, Shai Rosenstein, Miri Yavzori, Yael Dror, Ella Fudim, Bella Ungar, Uri Kopylov, et al. 2019. “Molecular Landscape of Anti-Drug Antibodies Reveals the Mechanism of the Immune Response Following Treatment With TNFα Antagonists.” Frontiers in Immunology 10 (December): 2921.